清华大学交叉信息院曾坚阳研究组成功开发了基于FISH数据和Hi-C数据的三维基因组重构方法,相关研究论文《基于FISH和Hi-C数据整合的三维基因组结构建模》(Integrating Hi-C and FISH data for modeling of the 3D organization of chromosomes)5月3日于《自然·通讯》在线发表。
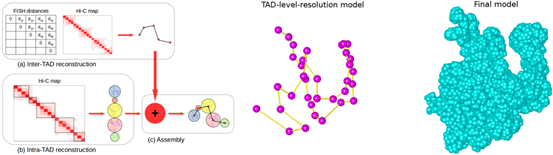
图:三维基因组结构建模的流程图
大量研究表明,染色体的空间结构与细胞内大多数的生物过程包括基因调控、DNA复制以及染色体异位等密切相关。因此,三维基因组结构解析对于细胞内生物过程的理解具有十分重要的意义。近年来,染色体构象捕获(3C)技术高速发展,通过与高通量测序技术相结合(Hi-C),可以直接获得全基因组范围内染色体相互作用图谱。基于染色体相互作用图谱,可以分析出染色体的层次结构并且重构出三维空间结构,从而为下游的分析和实验提供有价值的线索,并且为染色体折叠机制和功能的研究提供帮助。目前,由于Hi-C实验的限制,高分辨的三维基因组结构的重构难度较大。
曾坚阳研究组在三维基因组结构的重构方面具有丰富的经验,先后开发出了基于贝叶斯推断和流形学习并结合染色体物理特性的建模方法,相关成果均发表在核心期刊《核酸研究》(Nucleic Acids Research)上,其中基于流形学习的GEM方法被《基因组蛋白质组与生物信息学报》(Genomics,Proteomics & Bioinformatics,简称GPB)评为2018年国内生物信息学十大进展之一。
此次曾坚阳研究组发表于《自然》子刊的方法基于上述流形学习的模型首次将FISH数据与Hi-C数据相结合,极大程度上提高了结构建模的准确性。在该方法中,建模过程分为三个主要的步骤:第一,利用FISH数据和Hi-C数据来确定拓扑结构域(TADs)的相对空间位置;第二,利用Hi-C数据提供的几何约束以及多聚体的生物物理性质来确定TADs内的结构;第三,将上述两步进行融合,调整优化后得到最终的三维基因组结构。在与之前方法的比较中,该方法将结构的平均相对误差降低了近一半,进一步的下游分析表明,该方法可以准确构建出染色体的基本层次结构。
论文第一作者是曾坚阳研究组的埃及籍博士后艾哈迈德·阿巴斯(Ahmed Abbas),通讯作者是曾坚阳副教授,合作者包括清华大学张奇伟教授和高军涛研究员等。研究成果得到国家自然科学基金等项目经费的支持。
论文链接:https://www.nature.com/articles/s41467-019-10005-6
