Single-cell sequencing technologies have developed rapidly and provide unprecedented opportunities for biologists to investigate cellular states. However, it also poses new challenges in the form of experimental noise not found in bulk sequencing data, which might significantly decrease the accuracy of downstream bioinformatics analysis by introducing biases in the gene expression.
Now a team of Jianyang Zeng, Associate Professor at IIIS, Tsinghua University, along with teams from Peking University, Salk Institute for Biological Studies, University of California, and Shanghai Jiao Tong University, have proposed a new deep learning model, DeepSEM that would increase the accuracy of bioinformatics analysis in a paper, entitled “Modeling Gene Regulatory Networks Using Neural Network Architectures”, in Nature Computational Science.

Gene regulatory networks (GRNs) encode the complex molecular interactions that govern cell identity. In the paper, the team propose DeepSEM, a deep generative model that can jointly infer GRNs and biologically meaningful representation of single-cell RNA sequencing (scRNA-seq) data. In particular, a neural network version of the structural equation model (SEM) is developed to explicitly model the regulatory relationships among genes. Benchmark results show that DeepSEM achieves comparable or better performance on a variety of single-cell computational tasks, such as GRN inference, scRNA-seq data visualization, clustering and simulation, compared with the state-of-the-art methods. DeepSEM can provide a useful and powerful tool to analyze scRNA-seq data and infer a GRN.
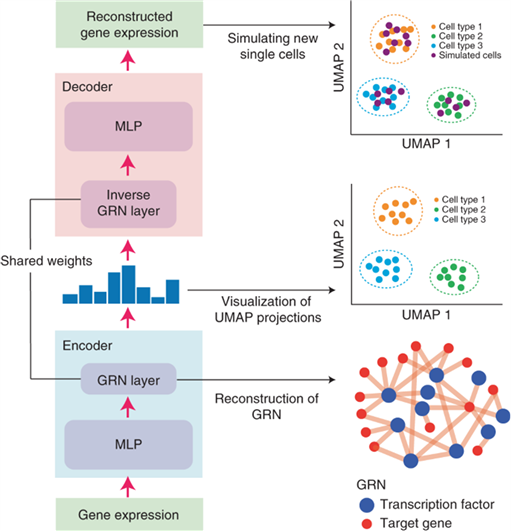
Overview of DeepSEM
In addition, DeepSEM may have other potential applications in single-cell biology, e.g., it can be adopted to integrate different single-cell modalities by leveraging GRN as a ‘bridge’ to construct a common latent space, and can be used to integrate other molecular interaction networks, such as a protein–protein interaction network, open chromatin data, DNA binding motifs and a genetic interaction network to further infer a GRN and achieve higher accuracy.
The first author of the paper is Hantao Shu, IIIS PhD student, and the co-correspondence authors are Jianzhu Ma, Associate Professor at Peking University, and Jianyang Zeng, Associate Professor at IIIS, Tsinghua University. This work was supported by National Natural Science Foundation of China and Nanjing Turing AI Institute.
The full paper is available at https://www.nature.com/articles/s43588-021-00099-8
