近日,清华大学交叉信息研究院曾坚阳课题组成功开发了一个基于序列的多尺度预测多肽和蛋白质相互作用的深度学习模型。该研究为多肽和蛋白质相互作用的机制提供了一个高效的预测框架,可以在为多肽药物预测结合靶点的同时,识别多肽序列上的结合位点。相关成果《一个多层次多肽-蛋白相互作用预测的深度学习框架》(A deep-learning framework for multi-level peptide–protein interaction prediction)于9月15日在《自然-通讯》(Nature Communications)上发表。

多肽和蛋白质的相互作用在生物体内起到关键的作用,参与多种细胞过程,比如信号传导、基因表达调控、细胞增殖和凋亡。识别和解析多肽和蛋白质的相互作用及其机制,有助于为多肽药物精准定位靶点,并为多肽药物的化学修饰提供信息,从而加速多肽药物的研发进程。目前有主流的计算框架分别基于序列的和基于结构来识别蛋白质和多肽配体的相互作用。然而,这些方法主要集中于识别蛋白质表面与多肽结合结合的残基,无法直接提取多肽序列中的结合残基。此外,基于结构的方法需要用到三维结构信息,但通过传统的实验方法测定得到的蛋白质-多肽复合物的结构非常昂贵且耗时。在本项研究中,作者提出了名为CAMP的深度学习框架,用于同时预测多肽-蛋白相互作用(PepPIs)和识别多肽序列上的结合残基。
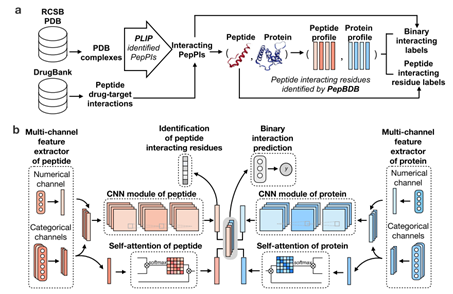
CAMP的模型框架图
CAMP将蛋白质和多肽的氨基酸序列、二级结构、理化性质、序列灵活性得分和蛋白质的PSSM矩阵作为模型输入,利用卷积神经网络(CNN)模块和自注意力机制(self-attention)来预测给定的肽-蛋白对之间是否存在相互作用,同时识别多肽序列上的结合位点。 实验结果表明,在多种数据划分的设定下,CAMP在基准数据集上的表现均优于现有的方法。此外,测试结果和案例分析表明,CAMP可以准确地预测多肽序列上的结合残基,从而为进一步理解多肽与蛋白质的结合机制提供有效的帮助。最后,作者进一步研究了CAMP在三个相关任务中的应用潜力,即多肽-蛋白结合域相互作用预测(peptide-PBD interaction)、结合亲和力评估和多肽的虚拟筛选。结果表明,CAMP在这三个相关任务上均获得出色表现。
综上所述,作者开发了一个多层次的多肽-蛋白相互作用预测的深度学习框架(CAMP)以同时预测多肽和蛋白质之间时候存在相互作用和识别多肽序列的结合残基。该方法使用多通道特征提取器分别处理数值特征和分类特征,以避免多源特征的不一致性。此外,作者通过和现有的最有方法比较,验证了CAMP拥有更优的性能,同时展示了CAMP在peptide-PBD相互作用预测、多肽-蛋白质的亲和力评估和多肽的虚拟筛选方面的应用潜力。这些结果表明,CAMP可以提供准确的肽-蛋白相互作用预测,并有助于研究者理解多肽与蛋白质结合的潜在机制。
该论文通讯作者为清华大学交叉信息研究院的曾坚阳副教授和赵诞助理研究员,第一作者为清华大学交叉信息研究院预研生雷逸品,合作作者包括清华大学自动化系李梢课题组等。此项研究工作获得国家自然科学基金和南京图灵人工智能研究院支持。
论文链接:
https://www.nature.com/articles/s41467-021-25772-4
